|
|

|

|
| e-Pub |
Section: Research Program
3D interaction and structure prediction
Participant : Amélie Héliou.
The biological function of macromolecules such as proteins and nucleic acids relies on their dynamic structural nature and their ability to interact with many different partners. This is specially challenging as structure flexibility is key and multi-scale modelling [19], [27] and efficient code are essential [32].
Our project covers various aspects of biological macromolecule structure and interaction modelling and analysis. First protein structure prediction is addressed through combinatorics. The dynamics of these types of structures is also studied using statistical and robotics inspired strategies. Both provide a good starting point to perform 3D interaction modelling, accurate structure and dynamics being essential.
Our group benefits from a good collaboration network, mainly at Stanford University (USA), Hkust (Hong-Kong) and McGill (Canada). The computational expertise in this field of computational structural biology is represented in a few large groups in the world (e.g. Pande lab at Stanford, Baker lab at U.Washington) that have both dry and wet labs. At Inria, our interest for structural biology is shared by the Abs and Orpailleur project-teams. Our activities are however now more centered around protein-nucleic acid interactions, multi-scale analysis, robotics inspired strategies and machine learning than protein-protein interactions, algorithms and geometry. We also shared a common interest for large biomolecules and their dynamics with the Nano-D project team and their adaptative sampling strategy. As a whole, we contribute to the development of geometric and machine learning strategies for macromolecular docking.
Game theory was used by M. Boudard in her PhD thesis, defended in 2015, to predict the 3d structure of RNA. In her PhD thesis, co-advised by J. Cohen (Lri ), A. Héliou is extending the approach to predict protein structures.
Robotics-inspired structure and dynamics
Participant : Amélie Héliou.
We recently work one a robotics approach to sample the conformational space of macromolecules like RNAs [1]. The robotics approach allows maintaining the secondary structure of the RNA fixed, as an unfolding is very unlikely and energetically demanding. By this approach we also dramatically reduce the number of degrees of freedom in the molecule. The conformational space becomes possible to be sampled. This reduction does not reduce the quality of the sampling.
|
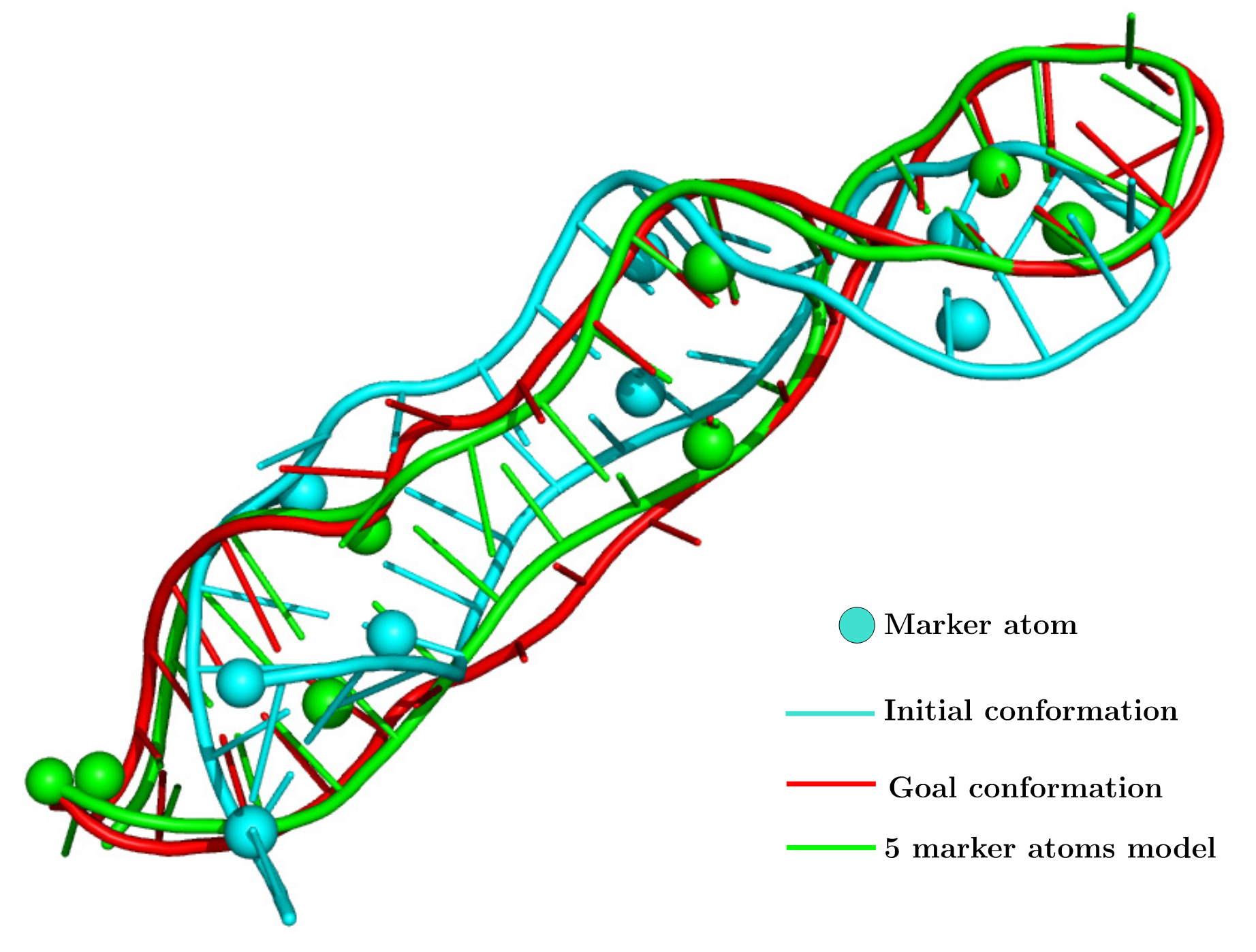
Our current work consists in applying the same approach to a targeted move. The motion is then driven either by the position of a few atoms or the distances between couple of atoms. Theses two aspects are under development and will increase the analysis possibility of experimental data. Our method can drive a RNA conformation toward another conformation of the same RNA given only the position of a few atoms (marker atoms).
For instance double electron-electron resonance (DEER) experimental results are distributions of distances. Probes are attached to the molecules and the distances between to probes is measured and outputted as a distribution. Our method is able to sample an ensemble of all-atom conformations that can explain the distance distribution.
Game theory and protein folding
Participant : Amélie Héliou.
M.Boudard used game theory to sample folded conformations of RNA. We work in apply game theory to sample folded conformations of proteins. This is challenging as a protein is generally less flexible than a RNA and thus accept less conformations.
Our work is first to find an algorithm that can guarantee the convergence to an Nash equilibrium (a state were no player would increase his payoff by playing something different alone) and prove their convergence. At the same time, we are looking for efficient and biologically relevant ways of defining the game settings so that Nash equilibria correspond to folded states. One direction would be to draw a parallel between Nash equilibria and local minima of the kinetic landscape.